يحتفل العالم في شهر مايو من كل عام وتحديدا في الثامن من الشهر باليوم العالمي لمرض الثلاسيميا. وبين عام وآخر، تظهر انجازات علمية وطبية جديدة في هذا المجال. وأحدث التطورات يتمثل بموافقة هيئة الغذاء والدواء الأمريكية (FDA) على العلاج الجيني المعروف باسم betibeglogene autotemcel، أو beti-cel "بيتا – سيل" في أغسطس من العام الماضي باعتباره آمناً وفعالاً للمرضى البالغين والأطفال الذين يعانون من مرض الثلاسيميا بيتا، الذي يحتاج إلى نقل الدم بصورة مستمرة.
يساعد العلاج الجيني في إحداث تحول إيجابي كبير في حياة المرضى الذين يعانون من مرض الثلاسيميا بيتا - وهو اضطراب جيني في الدم - من خلال مساعدتهم في إيقاف أو تقليل اعتمادهم على عمليات نقل الدم
حياة طبيعية
يمثل هذا العلاج علامة فارقة في رحلة علاج المرضى المصابين بالثلاسيميا، إذ يستخدم العلاج الجيني الجديد الخلايا الجذعية المكوّنة للدم من المرضى أنفسهم لإنتاج خلايا دم حمراء أكثر صحة وعلاج اضطرابات الدم لديهم - أو على الأقل التقليل على نحو ملحوظ من عدد عمليات نقل الدم اللازمة لإدارة حالتهم. وبفضل العلاج الجيني، يمكننا تذليل العقبات والتحديات التي يواجهها مرضى الثلاسيميا ومنحهم القدرة على متابعة أهدافهم وعيش حياتهم بشكل طبيعي وألا يكونوا مقيدين بعمليات نقل الدم أو مضاعفات زيادة الحديد.
ومؤخرا، أعلن "مستشفى كليفلاند كلينك" في دولة الإمارات، عن كونه أحد المراكز القليلة في العالم المعتمدة لتقديم العلاج الجيني الجديد وعلى مساعدة المرضى من جميع أنحاء العالم.
خصوصية شرق أوسطية
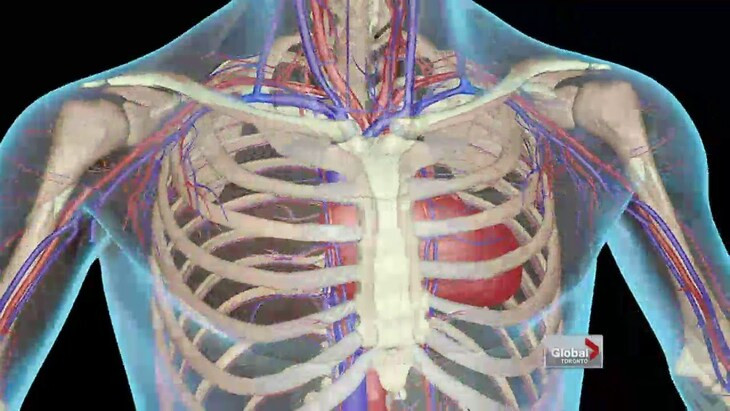
ثمة نوعان لمرض الثلاسيميا وهما ألفا وبيتا، ويُعد هذا المرض من أكثر الاضطرابات الجينية شيوعاً ويحدث بشكل متكرر لدى سكان مناطق الشرق الأوسط ودول البحر الأبيض المتوسط وشمال أفريقيا والهند وآسيا الوسطى وجنوب شرق آسيا.
ويؤثر مرض الثلاسيميا بيتا في قدرة الجسم على إنتاج الهيموجلوبين، وهو بروتين موجود في خلايا الدم الحمراء يسمح لها بنقل الأكسجين في جميع أنحاء الجسم. ويعاني الأفراد المصابون بمرض الثلاسيميا بيتا الكبرى بأعراض خطيرة في سن مبكرة، والتي يمكن علاجها عن طريق نقل الدم بشكل متكرر، ولكن عمليات نقل الدم نفسها تخلق خطر زيادة نسبة الحديد في الدم مع احتمال تضرر الكبد والقلب ونظام الغدد الصماء لدى المرضى.
ويقول الدكتور ربيع حنّا، اختصاصي أمراض الدم والأورام لدى الأطفال في مستشفى كليفلاند كلينك إن العلاج الجيني الجديد، الذي يساعد على هذه المشكلة، يُعد مثالياً للمرضى من المراهقين أو المرضى الأكبر سناً بقليل الذين بدؤوا يعانون بالفعل من مضاعفات نتيجة عمليات نقل الدم.
التوقيت المناسب
يوضح الدكتور حنّا أن السبب في كون هؤلاء المرضى مرشحين مثاليين لتلقي هذا النوع من العلاج يكمن في أن العلاج الجيني الجديد يشتمل على فترة قصيرة (أربعة أيام) من العلاج الكيميائي المسمّى "بوسولفان" والذي قد يؤثر في خصوبة المرضى الذكور والإناث، لذلك من الأفضل الانتظار حتى يصل المرضى سنوات المراهقة حتى يتمكنوا من الخضوع لعلاج الحفاظ على الخصوبة، رغم أن لدينا طرقاً للحفاظ على الخصوبة حتى لدى المرضى الذين لم يصلوا سن البلوغ.
من ناحية أخرى، من الأفضل عدم إهمال المرض لفترة طويلة لأنه بعد سنوات من الحقن، ربما تكون قلوب المرضى وأكبادهم قد تأثرت بشدة بالفعل بسبب فرط حمل الحديد في الجسم. وبشكل عام، كلما كان المرضى أكثر صحة في بداية عملية العلاج الجيني، كلما قلت الآثار الجانبية أقل.
آلية التعديل الجيني
ولفت الدكتور حنّا إلى أن علاج beti-cel "بيتا – سيل" ذو الحد الأدنى من التدخل الجراحي يتم على ثلاث مراحل على مدار عدة أشهر. يتم في المرحلة الأولى استخدام دواءين (G-CSF وplerixafor) لتحفيز نخاع عظم المريض على إنتاج المزيد من الخلايا الجذعية. وفي المرحلة الثانية، يتم سحب الدم لجمع الخلايا الجذعية من الدم المحيطي، ومن ثم يتم إرسال هذه الخلايا الجذعية إلى شركة العلاج لإجراء تعديل الجينات، أي إدخال جين يمكّنهم من إنتاج هيموجلوبين سليم غير متأثر. وتستغرق عملية التعديل حوالي شهرين، ليتم بعد ذلك إجراء المرحلة النهائية في المستشفى بحيث يمكن متابعة المرضى عن كثب، والذين سيتلقون علاجاً كيميائياً واحداً مرة واحدة يومياً لمدة أربعة أيام، تليها أيام راحة قليلة، لتهيئة الجسم لإعادة إدخال الخلايا الجذعية المعدّلة من خلال الحقن في الوريد، على غرار عملية الحقن الوريدي.
في غضون شهر تقريباً بعد ضخ الخلايا المعدّلة، تبدأ الخلايا الجذعية الجديدة لدى الغالبية العظمى من المرضى في العمل على نحو سليم ولن يحتاجوا إلى المزيد من عمليات نقل الدم أو العلاجات الأخرى، إذ سيكون لديهم خلايا مثبتة في نخاع العظام قادرة على إنتاج الهيموجلوبين بشكل طبيعي. وحتى في الحالات الاستثنائية القليلة التي لا يكون فيها العلاج الجيني فعّالاً بنسبة 100%، فإن حاجة المرضى لعمليات نقل الدم ستنخفض بشكل كبير.